Recipe 6: Error-trim reads using streaming k-mer abundance trimming¶
This is a recipe for trimming your reads at low-abundance k-mers, as
described in These are not the k-mers you are looking for: efficient
online k-mer counting using a probabilistic data structure. Unlike the
filter-abund script used in that paper, this approach does the
trimming in a streaming few-pass approach, in which most of the data
is only looked at once.
Low-abundance k-mer trimming is primarily useful for removing errors from short reads prior to assembly or mapping. This can significantly reduce memory requirements for assembly, in particular. However, note that you should only do this kind of error trimming in cases where your downstream analysis approaches won’t correct the errors for you; see On the optimal trimming of high-throughput mRNA sequence data, MacManes, 2014 for more information.
Note: at the moment, the khmer script trim-low-abund.py is in the
khmer repository under branch update/streaming. Once we’ve merged
it into the master branch and cut a release, we’ll remove this note
and simply specify the khmer release required.
If you have a collection of short reads, and you plot the k-mer abundance spectrum, you may note that there are many low-abundance k-mers – look at the origin on this plot
load-into-counting.py -x 1e8 -k 20 reads.kh reads.fa
abundance-dist.py -s reads.kh reads.fa reads.dist
./plot-abundance-dist.py reads.dist reads-dist.png --ymax=300
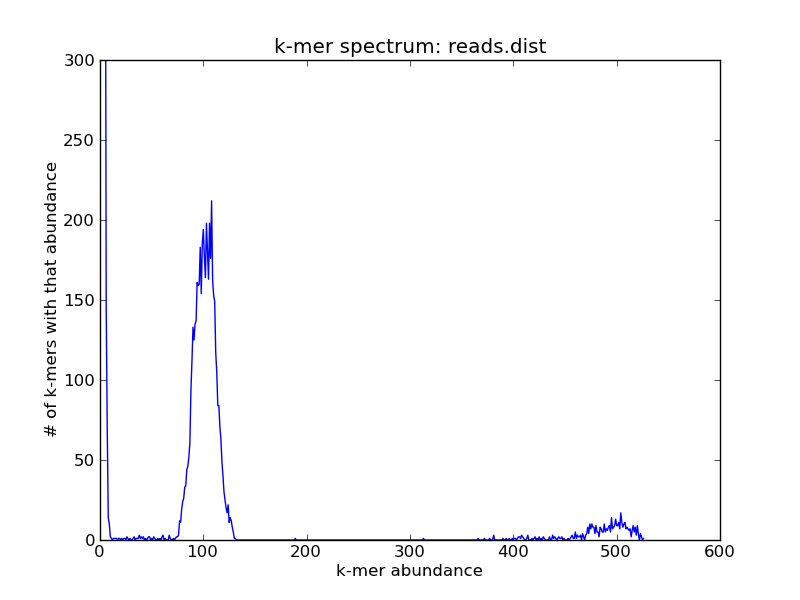
If you zoom in on the origin, you’ll see that in fact most of the k-mers in this sequence collection are unique
./plot-abundance-dist.py reads.dist reads-dist-2.png --xmax=20
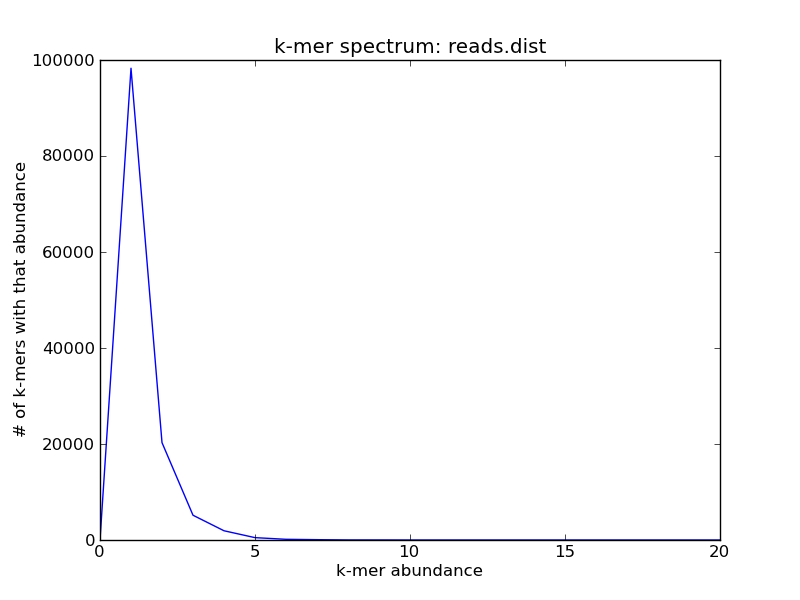
For high-coverage genomes, this will generally be due to sequencing errors; for variable coverage, this will be a mixture of real and erroneous k-mers.
You can use the sandbox script trim-low-abund.py to efficiently
trim sequences at these k-mers:
trim-low-abund.py -x 1e8 -k 20 reads.fa
(By default, trim-low-abund trims k-mers that are unique in reads that
have 20 or higher coverage. You can change the multiplicity of trimming
with -C and the trusted coverage with -Z.)
After running trim-low-abund, you’ll note that most of the unique k-mers are now gone:
load-into-counting.py -x 1e8 -k 20 reads-trim.kh reads.fa.abundtrim
abundance-dist.py -s reads-trim.kh reads.fa.abundtrim reads-trim.dist
./plot-abundance-dist.py reads-trim.dist reads-trim-dist.png --xmax=20 --ymax=90000
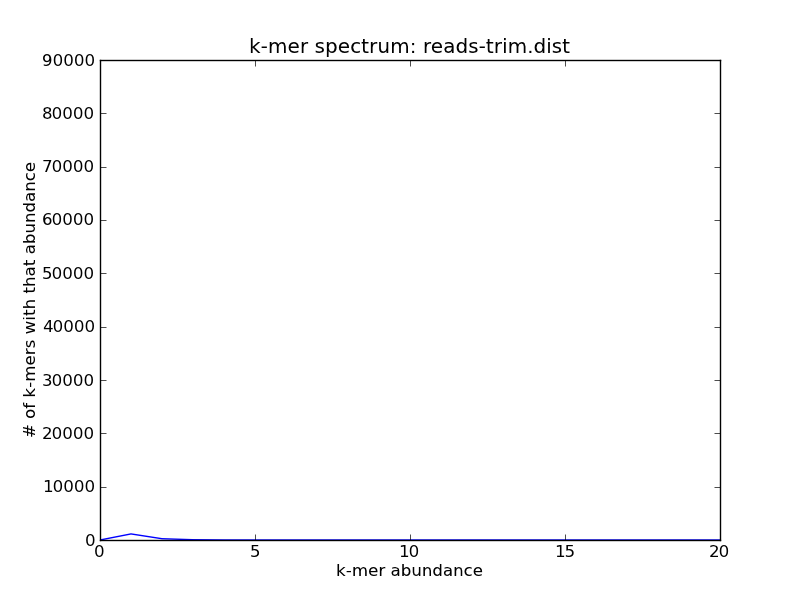
Voila!
As mentioned briefly above, here we are using a more memory- and time-
efficient approach than the filter-abund script that we published
as part of khmer. Note that you can use this script on metagenomes
and transcriptomes as well by passing in the -V parameter for
variable coverage trimming; this is discussed more in :doc:`../007-variable-coverage-trimming/index`__.
Resources and Links¶
This recipe is hosted in the khmer-recipes repository, https://github.com/ged-lab/khmer-recipes/.
It requires the khmer software.
LICENSE: This documentation and all textual/graphic site content is licensed under the Creative Commons - 0 License (CC0) -- fork @ github.
comments powered by Disqus