Recipe 5: Estimate genome size and coverage from shotgun sequencing data¶
In Recipe 1: Extract reads by coverage, we showed how to get a read coverage spectrum for a shotgun data set. This is a useful diagnostic tool that can be used to estimate total genome size, average coverage, and repetitive content.
Uses for this recipe include estimating the sequencing depth of a particular library, estimating the repeat content, and generally characterizing your shotgun data.
See Recipe 3: Estimate (meta)genome size from unassembled reads for a recipe that estimate the single-copy genome size; that recipe can be used for any metagenomic or genomic data set, including ones subjected to whole genome amplification, while this recipe can only be used for data from unamplified/unbiased whole genome shotgun sequencing.
Let’s assume you have a genome that you’ve sequenced to some unknown
coverage, and you want to estimate the total genome size, repeat
content, and sequencing coverage. If your reads are in reads.fa,
you can generate a read coverage spectrum from your genome like so:
load-into-counting.py -x 1e8 -k 20 reads.kh reads.fa
~/dev/khmer/sandbox/calc-median-distribution.py reads.kh reads.fa reads-cov.dist
./plot-coverage-dist.py reads-cov.dist reads-cov.png --xmax=600 --ymax=500
and the result looks like this:
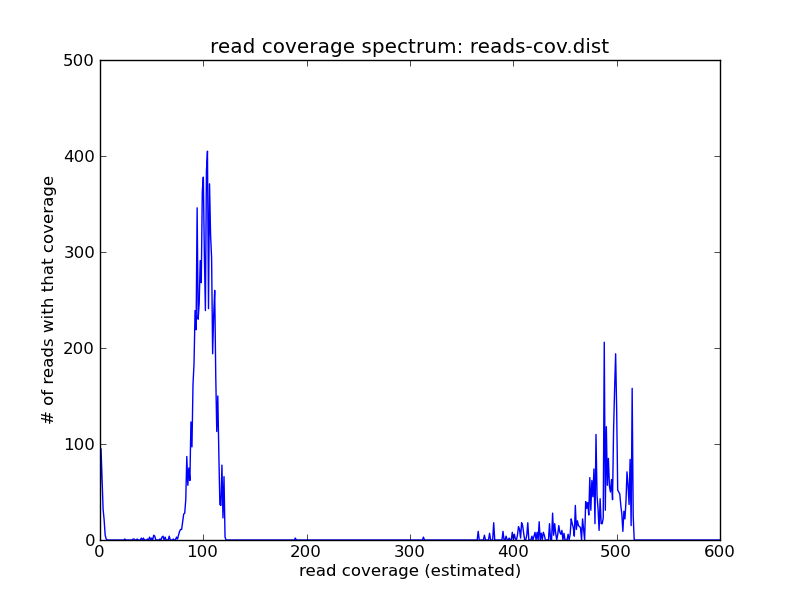
For this (simulated) data set, you can see three peaks: one on the far right, which contains the high-abundance reads from repeats; one in the middle, which contains the reads from the single-copy genome; and one all the way on the left at ~1, which contains all of the highly erroneous reads
Pick a coverage number near the peak that you think represents the single-copy genome - with sufficient coverage, and in the absence of significant heterozygosity, it should be the first peak that’s well to the right of zero. Here, we’ll pick 100.
Then run
./estimate-total-genome-size.py reads.fa reads-cov.dist 100
and you should see:
Estimated total genome size is: 7666 bp
Estimated size > 5x repeats is: 587 bp
Estimated single-genome coverage is: 109.8
The true values (for this simulated genome) are 7500bp genome size, 500 bp > 5x repeat, and 150x coverage, so this isn’t a terribly bad set of estimates. The single-genome coverage estimate is probably low because we’re estimating read coverage with a k-mer based approach, which is sensitive to highly erroneous reads.
A few notes:
- Yes, you could automate the selection of the first peak, but I think it’s always a good idea to look at your data!
- In the case of highly polymorphic diploid genomes, you would have to make sure to include the haploid reads but only count them 50% towards the final genome size.
Resources and Links¶
This recipe is hosted in the khmer-recipes repository, https://github.com/ged-lab/khmer-recipes/.
It requires the khmer software.
LICENSE: This documentation and all textual/graphic site content is licensed under the Creative Commons - 0 License (CC0) -- fork @ github.
comments powered by Disqus