Recipe 7: Trim metagenome and transcriptome reads with variable coverage k-mer trimming¶
This is a recipe for metagenome and transcriptome k-mer trimming. K-mer trimming for genomes (see Recipe 6: Error-trim reads using streaming k-mer abundance trimming) relies on using a hard cutoff to identify low-abundance k-mers that are likely to be erroneous in high coverage genomic shotgun sequencing data sets; however, some or many of these k-mers may not be errors in situations where you have a variety of molecular species at different abundances in your data set – specifically, when you are sequencing metagenomes or transcriptomes. (This will also work for whole-genome amplified data sets.)
Low-abundance k-mer trimming is primarily useful for removing errors from short reads prior to assembly or mapping. This can significantly reduce memory requirements for assembly, in particular. However, note that you should only do this kind of error trimming in cases where your downstream analysis approaches won’t correct the errors for you; see On the optimal trimming of high-throughput mRNA sequence data, MacManes, 2014 for more information.
Suppose you have a metagenome with several different coverage peaks; here, in this simulated data set, there are three: one at 10, one at 100, and one at about 300.
load-into-counting.py -x 1e8 -k 20 reads.kh reads.fa
~/dev/khmer/sandbox/calc-median-distribution.py reads.kh reads.fa reads-cov.dist
./plot-coverage-dist.py reads-cov.dist reads-cov.png --xmax=350
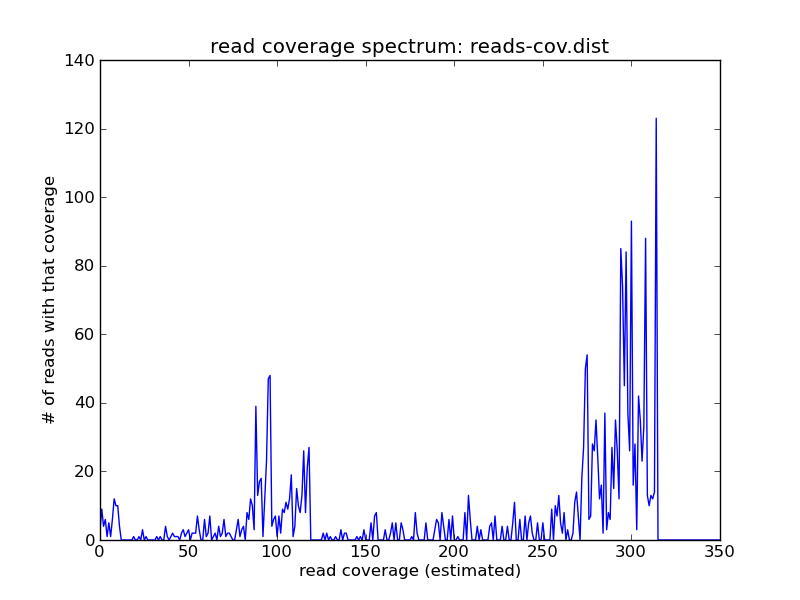
Suppose you wanted to remove errors with k-mer abundance trimming (as in Recipe 6: Error-trim reads using streaming k-mer abundance trimming) - the problem is that you can’t just use a hard cutoff, because some of the low-abundance k-mers are real, while some are not. For example, the k-mer spectrum of this data set is much broader at 1 than it would be for a high-coverage genome:
abundance-dist.py -s reads.kh reads.fa reads.dist
./plot-abundance-dist.py reads.dist reads-dist.png --xmax=20 --ymax=18000

You can use the -V argument to the script
trim-low-abund.py to efficiently trim sequences at low-abundance
k-mers that are in high-coverage reads. With -V, low-abundance k-mers
in low-coverage reads are kept; these are much more likely to be
correct than a low-abundance k-mer in a high-coverage read.
trim-low-abund.py -x 1e8 -k 20 -V reads.fa
(By default, trim-low-abund trims k-mers that are unique in reads that
have 20 or higher coverage. You can change the multiplicity of trimming
with -C and the trusted coverage with -Z.)
After running trim-low-abund, you’ll note that some but not all of the unique k-mers are now gone:
load-into-counting.py -x 1e8 -k 20 reads-trim.kh reads.fa.abundtrim
abundance-dist.py -s reads-trim.kh reads.fa.abundtrim reads-trim.dist
./plot-abundance-dist.py reads-trim.dist reads-trim-dist.png --xmax=20 --ymax=18000
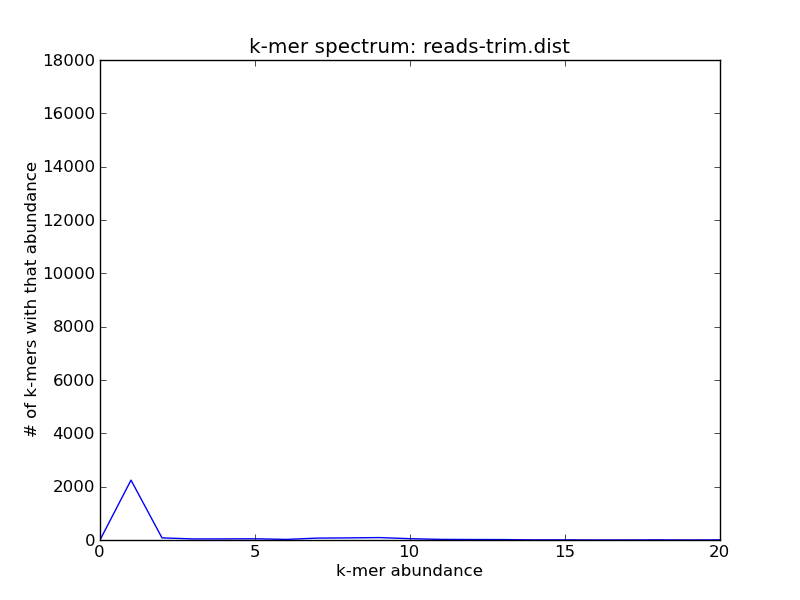
Voila!
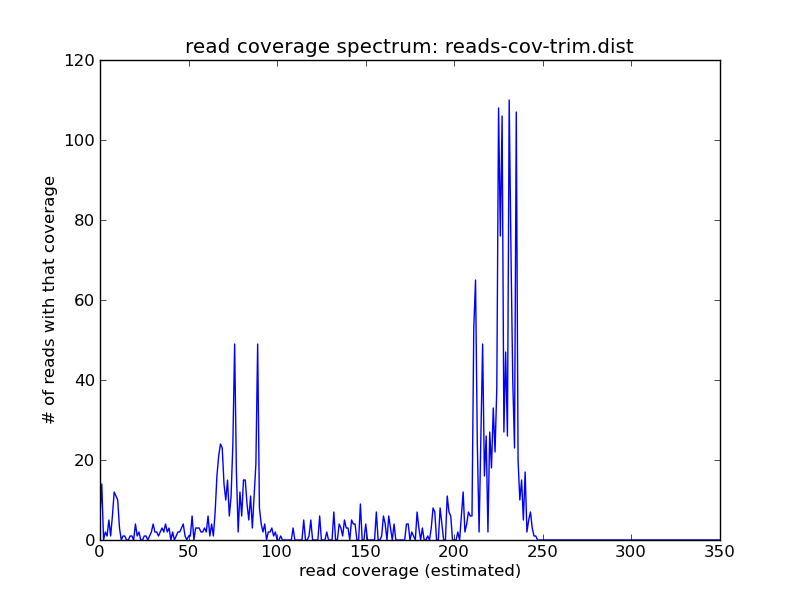
You can see that the abundance of the higher-coverage abundance has shifted, due to the trimming; but there are still reads at the 10x peak:
load-into-counting.py -x 1e8 -k 20 reads-trim.kh reads.fa.abundtrim
~/dev/khmer/sandbox/calc-median-distribution.py reads-trim.kh reads.fa.abundtrim reads-cov-trim.dist
./plot-coverage-dist.py reads-cov-trim.dist reads-cov-trim.png --xmax=350
Resources and Links¶
This recipe is hosted in the khmer-recipes repository, https://github.com/ged-lab/khmer-recipes/.
It requires the khmer software.
LICENSE: This documentation and all textual/graphic site content is licensed under the Creative Commons - 0 License (CC0) -- fork @ github.
comments powered by Disqus